In the intricate landscape of chemical understanding, few principles are as fundamental and yet as profoundly impactful as Hund’s Rule. Often encountered in introductory chemistry, its true significance blossoms when viewed through the lens of modern computational chemistry. This rule, originating from the work of Friedrich Hund, governs the filling of electrons within degenerate atomic and molecular orbitals, dictating the lowest energy state of an atom or molecule. While its conceptual elegance is undeniable, its practical application in predictive modeling and scientific software highlights its crucial role in the technological advancements that drive fields ranging from materials science to drug discovery. This article delves into Hund’s Rule not just as a theoretical concept, but as a critical parameter in the computational tools that scientists rely on daily.

The Fundamental Principle: Electron Configuration and Stability
At its core, Hund’s Rule provides a crucial insight into the electronic structure of atoms and molecules, directly impacting their stability and reactivity. Understanding this rule is the bedrock upon which many complex quantum mechanical calculations are built, especially within the technological domain of computational chemistry.
Degenerate Orbitals: The Playground for Electrons
Before delving into the rule itself, it’s essential to grasp the concept of degenerate orbitals. In atomic and molecular orbital theory, degenerate orbitals are those that possess the same energy level. For instance, within a given electron shell, the three p orbitals (px, py, pz) are degenerate. Similarly, the five d orbitals are degenerate. These orbitals can be visualized as regions of space around the nucleus where there is a high probability of finding an electron. When electrons are filling these orbitals, Hund’s Rule dictates the preferred arrangement.
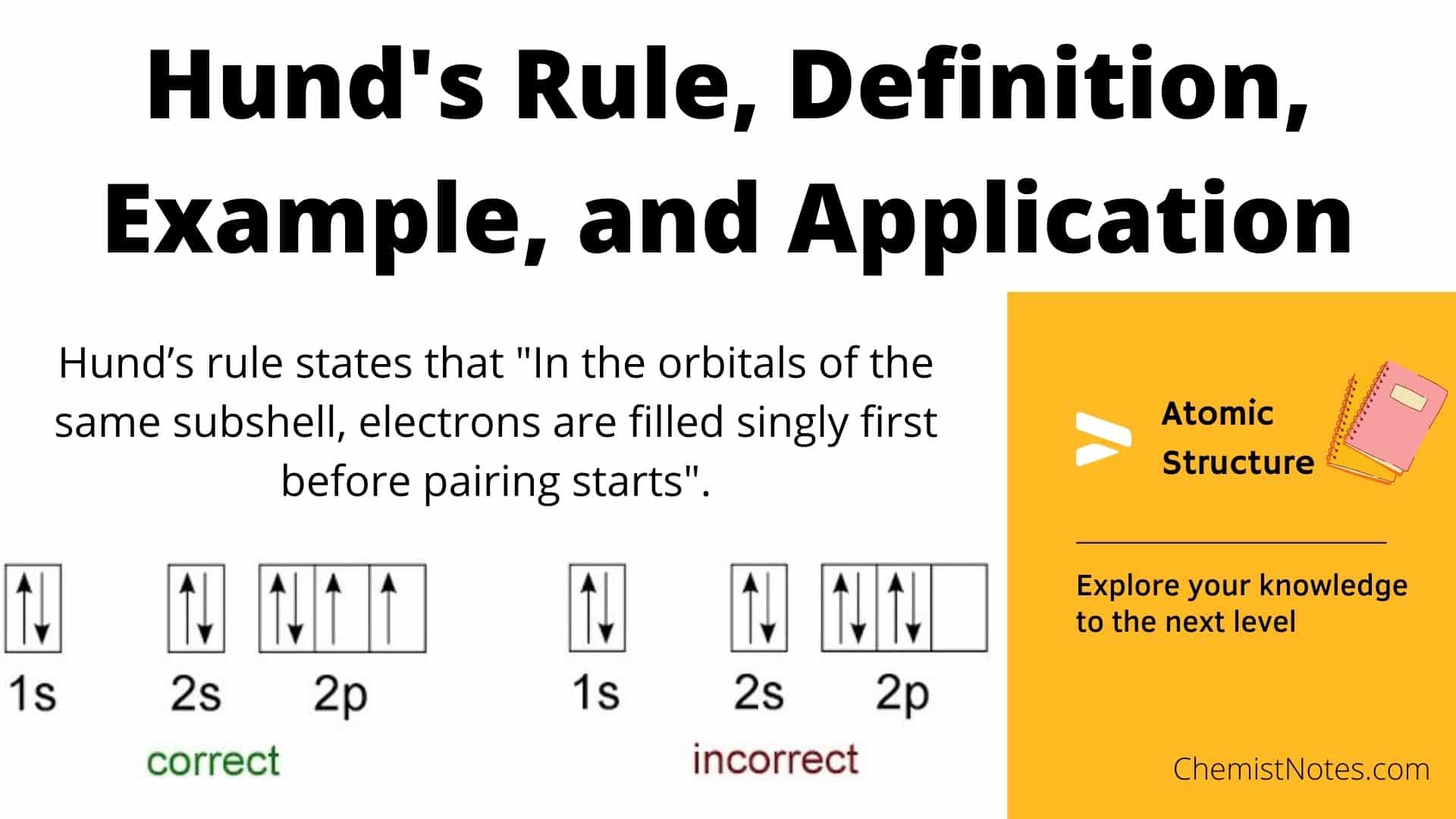
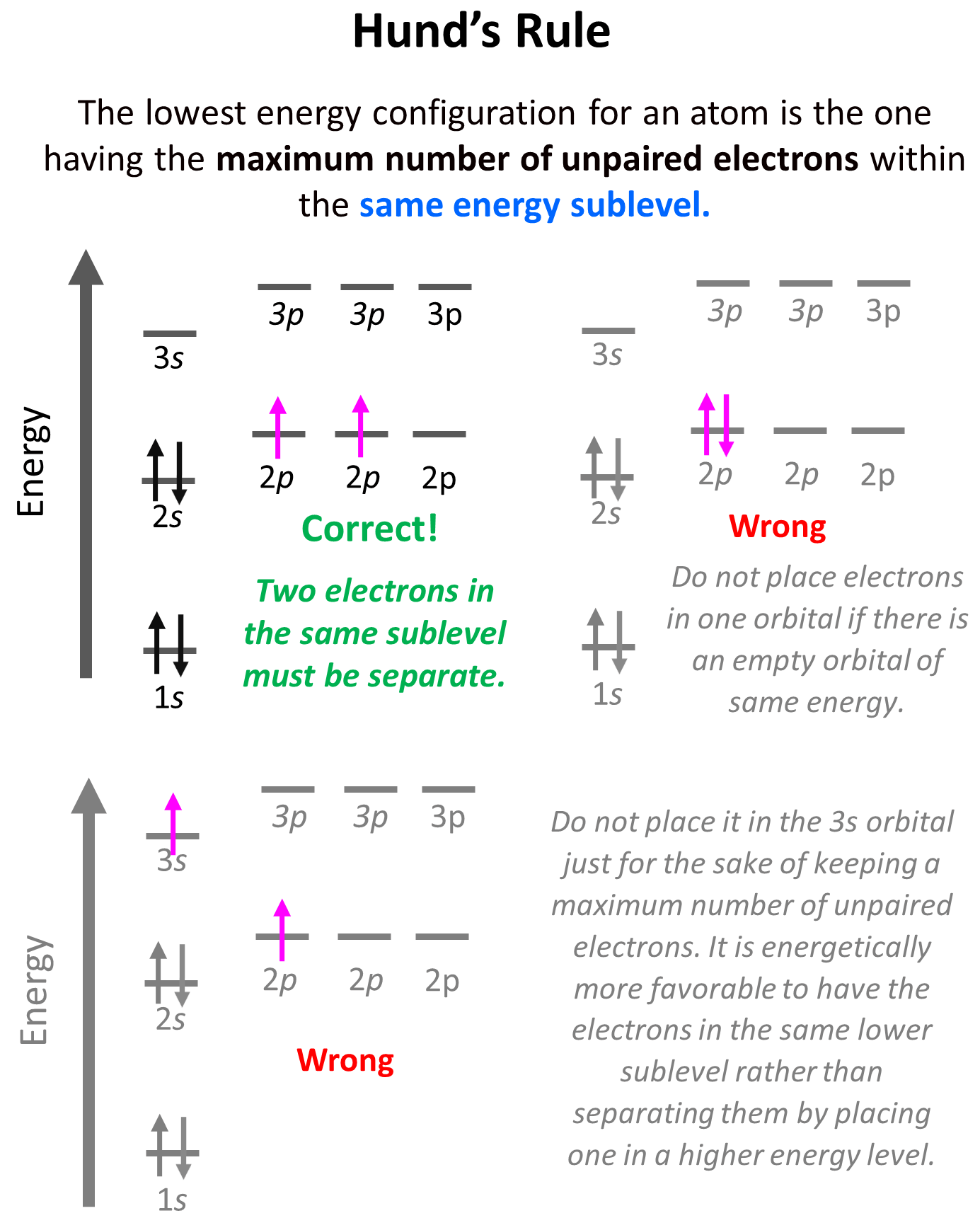
The Rule of Maximizing Spin Multiplicity
Hund’s Rule, in its most common formulation, states that for a given electron configuration, the term with the greatest spin multiplicity has the lowest energy. More simply, electrons will individually occupy each degenerate orbital before pairing up, and these single-occupied electrons will have parallel spins. This seemingly straightforward directive has profound implications for the stability of an electronic configuration.
Consider the filling of three degenerate p orbitals. If an atom has three electrons to place in these orbitals, Hund’s Rule dictates that each electron will go into a separate p orbital with its spin aligned in the same direction (e.g., all spin up). This arrangement maximizes the total spin of the atom, leading to a lower overall energy. If, instead, two electrons were paired in one orbital and the third occupied another, the total spin would be lower, and the energy higher. This preference for maximizing spin multiplicity stems from fundamental electrostatic repulsions and quantum mechanical exchange interactions. Electrons with parallel spins experience less repulsion and are thus in a more stable, lower-energy state.
Exchange Interaction: The Quantum Mechanical Driver
The underlying quantum mechanical phenomenon that drives Hund’s Rule is the exchange interaction. This is a non-classical effect that arises from the indistinguishability of electrons and the Pauli exclusion principle. When electrons have parallel spins, their wave functions are antisymmetrized in such a way that they tend to avoid each other more effectively than electrons with opposite spins. This reduction in electron-electron repulsion contributes to the stabilization of configurations with maximum spin.
Computational Chemistry: Implementing Hund’s Rule in Software
The theoretical underpinnings of Hund’s Rule are not merely academic curiosities; they are foundational algorithms and parameters integrated into sophisticated software packages used across various scientific and technological disciplines. Computational chemistry platforms leverage these rules to predict molecular properties, simulate chemical reactions, and design new materials.
Quantum Chemical Software: The Engine of Prediction
Modern computational chemistry relies on a suite of powerful software packages, such as Gaussian, Quantum ESPRESSO, ORCA, and GAMESS, to perform ab initio calculations (calculations from first principles) and density functional theory (DFT) simulations. These programs numerically solve the Schrödinger equation, or approximations thereof, to determine the electronic structure of molecules. Hund’s Rule, and its underlying principles, are intrinsically embedded within the algorithms that these software packages employ to determine ground-state electronic configurations.
When a user inputs a molecular structure, the software embarks on a complex process of approximating electron energies and wave functions. The algorithms for filling molecular orbitals, particularly in cases of degeneracy, must account for the energetic preferences described by Hund’s Rule. Failure to do so would result in inaccurate predictions of molecular stability, reactivity, and spectroscopic properties. This means that every time a computational chemist predicts the properties of a new molecule, Hund’s Rule is, in essence, being computationally enforced.
Basis Sets and Orbital Descriptions
The accuracy of computational chemistry calculations is heavily influenced by the “basis set” used – a set of mathematical functions that approximate atomic orbitals. Different basis sets offer varying levels of detail in describing the electron cloud. Within these descriptions, the handling of degenerate orbitals and the application of Hund’s Rule are critical. Software must correctly identify degenerate orbitals arising from the chosen basis set and molecular symmetry and then apply the filling rules dictated by Hund’s Rule to assign electrons and determine the lowest energy state. This involves sophisticated algorithms that can distinguish between different spin states and select the one with the greatest multiplicity.
Applications in Materials Science and Drug Discovery
The technological impact of accurately modeling electronic structure, guided by principles like Hund’s Rule, is vast. In materials science, researchers use computational tools to design novel materials with specific electronic or magnetic properties. For instance, understanding the electron filling in transition metal oxides, where d orbitals are heavily involved, is crucial for developing advanced catalysts, high-temperature superconductors, and magnetic storage media. Hund’s Rule plays a pivotal role in predicting the magnetic moments of these materials, which are directly related to the spin alignment of unpaired electrons.
In drug discovery, computational chemistry aids in understanding the interactions between drug molecules and biological targets. The electronic properties of molecules, including their charge distribution and orbital energies, influence their binding affinity and pharmacokinetic profiles. Accurate modeling, which implicitly incorporates Hund’s Rule for electron configurations, allows scientists to virtually screen potential drug candidates, saving significant time and resources compared to traditional experimental methods.
Beyond Simple Atoms: Hund’s Rule in Molecular Orbitals and Advanced Techniques
While Hund’s Rule is initially taught in the context of atomic orbitals, its principles extend to molecular orbitals, and its nuances are crucial for more advanced computational techniques.
Molecular Orbital Theory and Degeneracy
In molecular orbital (MO) theory, atomic orbitals combine to form molecular orbitals. Degenerate molecular orbitals can arise, particularly in molecules with high symmetry. For example, in certain aromatic systems, pi molecular orbitals can be degenerate. When filling these degenerate molecular orbitals with electrons, Hund’s Rule remains the guiding principle for achieving the lowest energy configuration. The software must identify these degenerate molecular orbitals and populate them with individual electrons of parallel spin before pairing them. This is essential for accurately predicting the electronic ground state and spectroscopic properties of molecules.
Spin Contamination and Multi-Reference Calculations
In some complex electronic systems, especially those with significant electron correlation or open shells (where electrons are unpaired), the simple single-determinant approximation used in basic Hartree-Fock or DFT calculations can lead to issues like “spin contamination.” This occurs when the calculated wave function possesses a spin state that is not the true ground state. Hund’s Rule, and the principles it represents, are implicitly addressed by more advanced computational methods designed to mitigate spin contamination.
Techniques such as Configuration Interaction (CI) or Multi-Configurational Self-Consistent Field (MCSCF) methods are employed to treat systems where a single electronic configuration is insufficient. These methods explicitly consider multiple electronic configurations and their interactions. While not a direct “rule” to be implemented, the fundamental tendency for electrons to minimize energy by maximizing spin multiplicity, as described by Hund’s Rule, is a guiding principle that these advanced methods aim to capture more rigorously. The software implementing these methods must understand the energetic landscape of various spin states, which is directly informed by Hund’s Rule.
Relativistic Effects and Electron Configurations
For heavy elements, relativistic effects become significant and can subtly influence electron configurations and orbital energies. While Hund’s Rule itself is derived from non-relativistic quantum mechanics, its application within relativistic computational schemes needs to account for these relativistic corrections. Modern quantum chemistry software packages designed for heavy elements incorporate relativistic Hamiltonians, and the filling of orbitals, still guided by the principles of minimizing energy and maximizing spin multiplicity, must be performed within this relativistic framework. This highlights how even fundamental rules are integrated and adapted within sophisticated technological tools to address increasingly complex chemical systems.
The Enduring Significance of Hund’s Rule in the Technological Age
From its humble beginnings as a rule for electron filling in introductory chemistry, Hund’s Rule has evolved into a critical component of the technological infrastructure that underpins modern scientific discovery. The ability of computational chemistry software to accurately implement this rule is not just about theoretical rigor; it is about enabling predictions that drive innovation.
Enabling Predictive Power in Software
The predictive power of computational chemistry is directly proportional to the accuracy of the underlying algorithms and the fundamental principles they embody. Hund’s Rule, by dictating the most stable electronic configurations, provides a crucial constraint that computational methods must adhere to. Without this adherence, simulations would produce erroneous results, rendering them useless for practical applications. The continuous development of more sophisticated algorithms and more powerful computing hardware allows for the ever-more-precise application of Hund’s Rule in solving complex chemical problems.
Facilitating Design and Discovery
The fields of materials science, pharmaceuticals, and catalysis are heavily reliant on the ability to design molecules and materials with specific properties. Hund’s Rule, as interpreted and implemented by computational tools, plays a vital role in this design process. By understanding how electrons will occupy orbitals and influence the overall electronic structure and spin states, researchers can rationally design new catalysts with enhanced activity, develop novel electronic materials with tailored conductivity, and engineer drug molecules with improved efficacy and reduced side effects.
The Future of Computational Chemistry and Hund’s Rule
As computational power continues to grow and quantum mechanical algorithms become more sophisticated, the integration of fundamental rules like Hund’s Rule will only become more refined. Future advancements may involve even more seamless integration of relativistic and electron correlation effects directly into the logic of how electrons fill orbitals. The ongoing evolution of artificial intelligence and machine learning in chemistry also promises new ways to leverage fundamental principles like Hund’s Rule, potentially leading to even faster and more accurate predictive models. In essence, Hund’s Rule remains a cornerstone, a foundational piece of knowledge that the technological marvels of computational chemistry build upon, ensuring that our understanding of the molecular world continues to expand and translate into tangible technological progress.
aViewFromTheCave is a participant in the Amazon Services LLC Associates Program, an affiliate advertising program designed to provide a means for sites to earn advertising fees by advertising and linking to Amazon.com. Amazon, the Amazon logo, AmazonSupply, and the AmazonSupply logo are trademarks of Amazon.com, Inc. or its affiliates. As an Amazon Associate we earn affiliate commissions from qualifying purchases.